

Transduction du signal et cibles thérapeutiques dans les maladies neurodégénératives
Transduction du Signal et Cibles Thérapeutiques dans les Maladies Neurodégénératives
L’équipe cherche à décrypter les mécanismes moléculaires et cellulaires de la mort neuronale dans plusieurs maladies neurodégénératives amyloïdes comme les maladies à prions, la maladie d’Alzheimer et d’autres pathologies du système nerveux central. Notre objectif est d’identifier des cibles (récepteurs, effecteurs de signalisation, microARN…) sur lesquelles il serait possible d’agir par des approches pharmacologiques ou génétiques pour limiter, voire enrayer, le processus neurodégénératif.
Nos travaux combinent des approches de biologie cellulaire, biologie moléculaire, biochimie, et d’imagerie afin d’identifier des voies de signalisation dérégulées par différentes protéines anormalement conformées (ou protéines amyloïdes) qui contribuent à la neurodégénérescence dans les maladies neurodégénératives amyloïdes.
Nos recherches s’articulent autour de trois axes :
Axe 1 : Identification de mécanismes de neurodégénérescence communs à plusieurs maladies neurodégénératives amyloïdes
Même si les maladies neurodégénératives amyloïdes ont des étiologies et des manifestations cliniques distinctes, la mort des neurones pourrait être régie par des mécanismes communs. Partant de l’étude des maladies à prions, nos travaux établissent pour la première fois que la dégénérescence des neurones dans les maladies à prions et la maladie d’Alzheimer impliquent les mêmes voies de signalisation (Pietri et al. Nat. Med. 2013). Ces voies de signalisation sont sous le contrôle d’un récepteur membranaire, la protéine prion cellulaire (PrPC), qui possède la propriété intrinsèque de fixer différentes protéines amyloïdes (prions pathogènes, peptides Ab de l’Alzheimer, a-synucléine de la maladie de Parkinson...) indépendamment de leur identité protéique. L’interaction de la PrPC avec les prions pathogènes ou les peptides Ab modifie la fonction de signalisation homéostatique de la PrPC, menant à l’activation chronique de plusieurs effecteurs de signalisation (i.e., les kinases ROCK, PDK1 et PDK4), ce qui a pour effet de rendre les neurones hypersensibles au stress inflammatoire, d’altérer la polarité neuronale (Alleaume-Butaux et al. Plos Pathogens 2015), d’amplifier la production des protéines amyloïdes (Ezpeleta et al. Nat Commun. 2019), ou encore de déréguler le métabolisme énergétique et l’équilibre redox des neurones (Arnould et al. Plos Pathogens 2021). Les kinases ROCK, PDK1 et PDK4 constituent aujourd’hui des cibles thérapeutiques potentielles pour lutter contre les maladies à prions ou la maladie d’Alzheimer.
Par des approches structure-fonction, nous cherchons actuellement à identifier les mécanismes menant à l’hyperactivation de ces effecteurs de signalisation, notamment de la kinase PDK1, dans les neurones malades et à cerner d’autres conséquences neuropathologiques associées à l’augmentation d’activité de ces effecteurs. Nos travaux visent également à objectiver si ces mêmes voies de signalisation dépendantes de la PrPC sont impliquées dans d’autres contextes neurodégénératifs, telles la sclérose latérale amyotrophique (maladie de Charcot), la maladie de Parkinson, ou les démences frontotemporales.
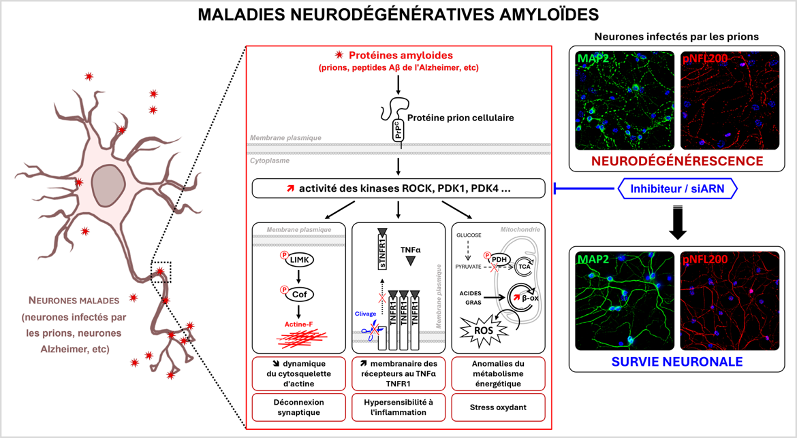
Effecteurs de signalisation et fonctions cellulaires contrôlés par la PrPC affectés par les protéines amyloïdes dans les neurones (infection à prions, neurones Alzheimer…). Clichés d’immunofluorescence montrant que l’inhibition de la kinase ROCK dans les cultures primaires de neurones en grain du cervelet infectés par les prions pathogènes protège les dendrites (marquage MAP2 en vert) et les axones (marquage pNFL200 en rouge) de la dégénérescence. Le noyau apparaît en bleu.
Axe 2 : Implication de la kinase PDK1 dans la neuroinflammation.
La neuroinflammation représente une composante importante du processus neurodégénératif, caractérisée par une production excessive et prolongée de facteurs inflammatoires tels la cytokine TNFa et les interleukines 1b et 6. Nos travaux en cours supportent un rôle de PDK1 dans la production de cytokines et chimiokines inflammatoires par les cellules immunitaires challengées avec des protéines amyloïdes, des agents bactériens ou viraux (Brevets WO2021/1911460 et WO2023/046941). Notre objectif est de cerner, sur le plan mécanistique, comment les protéines amyloïdes déclenchent la production des médiateurs de l’inflammation dans les cellules immunitaires (récepteurs et voies de signalisation impliqués) et comment PDK1 s’inscrit dans les voies de signalisation pro-inflammatoires.
Axe 3 : Rôle de la matière nanoparticulaire dans l’induction/progression de la maladie d’Alzheimer.
Dans la très grande majorité des cas, les individus atteints par la maladie d’Alzheimer sont sporadiques, c’est-à-dire qu’aucune anomalie génétique ne peut expliquer la pathologie. Un rôle de l’environnement est suspecté contribuer à l’émergence de cette maladie. L’exposition à la composante nanoparticulaire de la pollution atmosphérique est d’ailleurs considérée comme être un facteur de risque pour la maladie d’Alzheimer. Nos travaux démontrent qu’une exposition de neurones en culture ou de souris à des nanoparticules de dioxyde de titane ou de noir de carbone, des nanoparticules manufacturées fréquemment retrouvées dans notre environnement, induit des signes moléculaires évocateurs de la maladie d’Alzheimer comme une surproduction de peptides Ab (Ribeiro et al. Particle and Fibre Toxicology, 2022), apportant une base mécanistique quant au rôle de la matière nanoparticulaire dans l’étiologie de la maladie d’Alzheimer. Notre objectif est de déterminer si ces nanoparticules contribuent aussi à la progression de la maladie en influençant la fibrillation des peptides Ab et le dépôt de ces peptides sous la forme de plaques amyloïdes dans le cerveau.
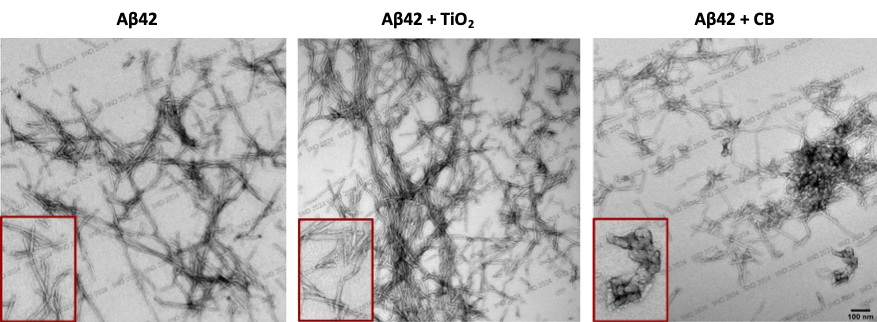
Images de microscopie électronique à transmission des peptides amyloïdes-β42 (Aβ42) incubés 96h en présence ou non de nanoparticules de dioxyde de titane (TiO2) ou de noir de carbone (CB). Alors que les nanoparticules de TiO2 semblent amplifier la formation de fibrilles de peptides Aβ42, les nanoparticules de CB induisent des agrégats amorphes de peptides.
Membres de l’équipe :
Benoit Schneider, DR1 CNRS, Pr Ecole polytechnique
Anne Baudry, CRCN INSERM
Mathéa Pietri, MCU-HC Université Paris Cité
Aurélie Alleaume-Butaux, IR Ecole polytechnique
Marc Dauplais, IR Ecole polytechnique
Chloé Bizingre, stagiaire post-doctorante
Flavien Picard, stagiaire post-doctorant
Clara Bianchi, étudiante en thèse
Florence Roussel, étudiante en thèse
Publications :
Bizingre C, Bianchi C, Baudry A, Alleaume-Butaux A, Schneider B, Pietri M. Post-translational modifications in prion diseases. Front Mol Neurosci. 2024 Jul 1;17:1405415. doi: 10.3389/fnmol.2024.1405415.
Ribeiro LW, Pietri M, Ardila-Osorio H, Baudry A, Boudet-Devaud F, Bizingre C, Arellano-Anaya ZE, Haeberlé AM, Gadot N, Boland S, Devineau S, Bailly Y, Kellermann O, Bencsik A, Schneider B. Titanium dioxide and carbon black nanoparticles disrupt neuronal homeostasis via excessive activation of cellular prion protein signaling. Part Fibre Toxicol. 2022 Jul 15;19(1):48. doi: 10.1186/s12989-022-00490-x.
Arnould H, Baudouin V, Baudry A, Ribeiro LW, Ardila-Osorio H, Pietri M, Caradeuc C, Soultawi C, Williams D, Alvarez M, Crozet C, Djouadi F, Laforge M, Bertho G, Kellermann O, Launay JM, Schmitt-Ulms G, Schneider B. Loss of prion protein control of glucose metabolism promotes neurodegeneration in model of prion diseases. PLoS Pathog. 2021 Oct 5;17(10):e1009991. doi: 10.1371/journal.ppat.1009991. eCollection 2021 Oct.
Schneider B, Baudry A, Pietri M, Alleaume-Butaux A, Bizingre C, Nioche P, Kellermann O, Launay JM. The Cellular Prion Protein-ROCK Connection: Contribution to Neuronal Homeostasis and Neurodegenerative Diseases. Front Cell Neurosci. 2021 Apr 12;15:660683. doi: 10.3389/fncel.2021.660683. eCollection 2021.
Ezpeleta J, Baudouin V, Arellano-Anaya ZE, Boudet-Devaud F, Pietri M, Baudry A, Haeberlé AM, Bailly Y, Kellermann O, Launay JM, Schneider B. Production of seedable Amyloid-beta peptides in model of prion diseases upon PrP(Sc)-induced PDK1 overactivation. Nat Commun. 2019 Aug 1;10(1):3442. doi: 10.1038/s41467-019-11333-3.
Alleaume-Butaux A, Nicot S, Pietri M, Baudry A, Dakowski C, Tixador P, Ardila-Osorio H, Haeberlé AM, Bailly Y, Peyrin JM, Launay JM, Kellermann O, Schneider B. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog. 2015 Aug 4;11(8):e1005073. doi: 10.1371/journal.ppat.1005073. eCollection 2015 Aug.
Pietri M, Dakowski C, Hannaoui S, Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A, Mouillet-Richard S, Haik S, Bailly Y, Peyrin JM, Launay JM, Kellermann O, Schneider B. PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer's diseases. Nat Med. 2013 Sep;19(9):1124-31. doi: 10.1038/nm.3302.