

Signal Transduction and Therapeutic Targets in Neurodegenerative Diseases
The team is seeking to decipher the molecular and cellular mechanisms underlying neuronal death in several amyloid-based neurodegenerative diseases such as prion diseases, Alzheimer’s disease, and other pathologies of the central nervous system. Our objective is to identify targets (receptors, signaling effectors, microRNAs...) on which it would be possible to act using pharmacological or genetic tools to limit or even stop the neurodegenerative process.
Our work combines approaches from cell biology, molecular biology, biochemistry and imaging to identify signaling pathways deregulated by distinct abnormally folded proteins (aka amyloid proteins) involved in neurodegeneration.
We develop three main axes:
Axis 1: Identification of neurodegenerative mechanisms common to amyloid-based neurodegenerative diseases
Although neurodegenerative diseases display distinct etiologies and clinical manifestations, neuronal death may be governed by common mechanisms. Starting from the study of prion diseases, our work provided prime evidence that neurodegeneration relies on the same signaling pathways in prion diseases and in Alzheimer’s disease (Pietri et al. Nat. Med. 2013). These signaling pathways are under the control of a membrane receptor, the cellular prion protein (PrPC), which displays the intrinsic property to bind several unrelated amyloid proteins (pathogenic prions, Alzheimer’s Ab peptides, Parkinson’s a-synuclein...) regardless of their protein identity. The interaction of PrPC with pathogenic prions or Ab peptides corrupts the homeostatic PrPC signaling function leading to the chronic activation of several signaling effectors (i.e., ROCK, PDK1 and PDK4 kinases), which render neurons hypersensitive to inflammatory stress, alter neuronal polarity (Alleaume-Butaux et al. Plos Pathogens 2015), amplify the production of neurotoxic amyloid proteins (Ezpeleta et al. Nat Common. 2019), or deregulate the energy metabolism and redox balance of neurons (Arnould et al. Plos Pathogens 2021). The ROCK, PDK1 and PDK4 kinases are today promising therapeutic targets to fight against prion diseases or Alzheimer’s disease.
By structure-function approaches, we currently seek to identify the mechanisms leading to the overactivation of these signaling effectors, notably the PDK1 kinase, in diseased neurons and to identify other neuropathological consequences associated with increased activity of these effectors. Our work also aims to assess whether these same PrPC-coupled signaling pathways are involved in other neurodegenerative contexts, such as amyotrophic lateral sclerosis (Charcot disease), Parkinson’s disease, or frontotemporal dementias.
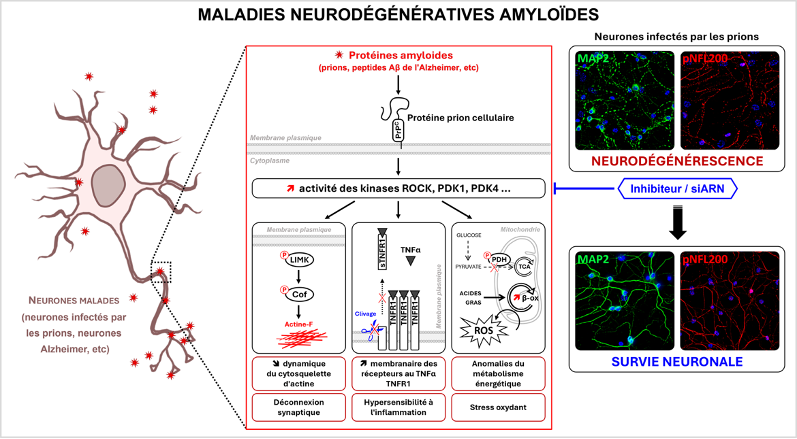
PrPC-coupled signaling effectors and cell functions altered in diseased neurons (prion infected neurons, Alzheimer’s neurons…). Immunofluorescence experiments showing that inhibition of ROCK in primary cerebellar granule neurons infected with prions protects dendrites (MAP2 staining in green) and axons (pNFL200 staining in red) from degeneration. The nucleus is in blue.
Axis 2: Involvement of PDK1 kinase in neuroinflammation.
Neuroinflammation is an important component of the neurodegenerative process characterized by excessive and prolonged production of inflammatory factors such as the cytokine TNFa and the interleukins 1 and 6. Our current work supports a role of PDK1 in the production of inflammatory factors by immune cells challenged with amyloid proteins, bacterial or viral agents (Patents WO/2021/1911460 and WO2023/046941). Our objective is to identify at a mechanistic level how amyloid proteins trigger the production of inflammatory factors in immune cells (receptors and signaling pathways involved) and how PDK1 fits into the pro-inflammatory signaling pathways.
Axis 3: Role of nanoparticle matter in the induction/progression of Alzheimer’s disease.
In the vast majority of cases, individuals with Alzheimer’s disease are sporadic, meaning that no genetic abnormalities can explain the pathology. It is suspected that the environment contributes to the emergence of this disease. Brain exposure to the nanoparticle component of air pollution is today considered a risk factor for Alzheimer’s disease. Our work shows that exposure of cultured neurons or mice to titanium dioxide or carbon black nanoparticles, two manufactured nanoparticles often found in our environment, induces molecular signs of Alzheimer’s disease, including overproduction of Ab peptides (Ribeiro et al. Particle and Fibre Toxicology, 2022). Our results provide a mechanistic basis for the etiological role of nanoparticle matter in the etiology of Alzheimer’s disease. Our objective is to determine whether these nanoparticles also contribute to the progression of the disease by influencing the fibrillation of Ab peptides and brain deposition of these peptides as amyloid plaques.
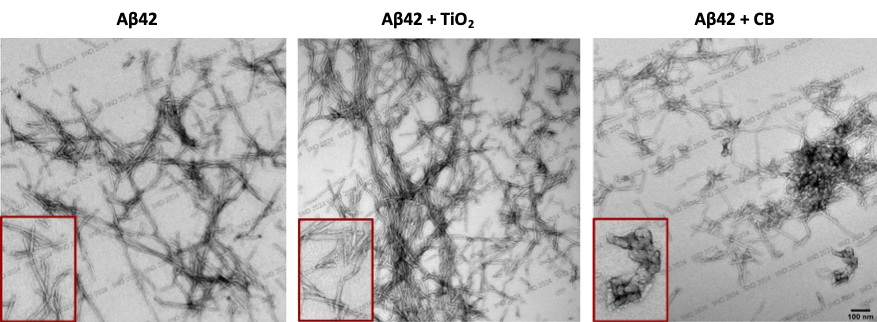
Transmission electron micrographs of negatively stained-amyloid-β 42 (Aβ 42) peptides incubated for 96h with or without nanoparticles of titanium dioxide (TiO2) or carbon black (CB). While TiO2 nanoparticles seem to amplify the production of Aβ fibrils, CB nanoparticles generate amorphous aggregates of Aβ peptides.
Team members:
Benoit Schneider, DR1 CNRS, Pr Ecole polytechnique
Anne Baudry, CRCN INSERM
Mathéa Pietri, MCU-HC Université Paris Cité
Aurélie Alleaume-Butaux, IR Ecole polytechnique
Marc Dauplais, IR Ecole polytechnique
Chloé Bizingre, post-doctoral fellow
Flavien Picard, post-doctoral fellow
Clara Bianchi, Ph.D. student
Florence Roussel, Ph.D. student
Selected publications :
Bizingre C, Bianchi C, Baudry A, Alleaume-Butaux A, Schneider B, Pietri M. Post-translational modifications in prion diseases. Front Mol Neurosci. 2024 Jul 1;17:1405415. doi: 10.3389/fnmol.2024.1405415.
Ribeiro LW, Pietri M, Ardila-Osorio H, Baudry A, Boudet-Devaud F, Bizingre C, Arellano-Anaya ZE, Haeberlé AM, Gadot N, Boland S, Devineau S, Bailly Y, Kellermann O, Bencsik A, Schneider B. Titanium dioxide and carbon black nanoparticles disrupt neuronal homeostasis via excessive activation of cellular prion protein signaling. Part Fibre Toxicol. 2022 Jul 15;19(1):48. doi: 10.1186/s12989-022-00490-x.
Arnould H, Baudouin V, Baudry A, Ribeiro LW, Ardila-Osorio H, Pietri M, Caradeuc C, Soultawi C, Williams D, Alvarez M, Crozet C, Djouadi F, Laforge M, Bertho G, Kellermann O, Launay JM, Schmitt-Ulms G, Schneider B. Loss of prion protein control of glucose metabolism promotes neurodegeneration in model of prion diseases. PLoS Pathog. 2021 Oct 5;17(10):e1009991. doi: 10.1371/journal.ppat.1009991. eCollection 2021 Oct.
Schneider B, Baudry A, Pietri M, Alleaume-Butaux A, Bizingre C, Nioche P, Kellermann O, Launay JM. The Cellular Prion Protein-ROCK Connection: Contribution to Neuronal Homeostasis and Neurodegenerative Diseases. Front Cell Neurosci. 2021 Apr 12;15:660683. doi: 10.3389/fncel.2021.660683. eCollection 2021.
Ezpeleta J, Baudouin V, Arellano-Anaya ZE, Boudet-Devaud F, Pietri M, Baudry A, Haeberlé AM, Bailly Y, Kellermann O, Launay JM, Schneider B. Production of seedable Amyloid-beta peptides in model of prion diseases upon PrP(Sc)-induced PDK1 overactivation. Nat Commun. 2019 Aug 1;10(1):3442. doi: 10.1038/s41467-019-11333-3.
Alleaume-Butaux A, Nicot S, Pietri M, Baudry A, Dakowski C, Tixador P, Ardila-Osorio H, Haeberlé AM, Bailly Y, Peyrin JM, Launay JM, Kellermann O, Schneider B. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog. 2015 Aug 4;11(8):e1005073. doi: 10.1371/journal.ppat.1005073. eCollection 2015 Aug.
Pietri M, Dakowski C, Hannaoui S, Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A, Mouillet-Richard S, Haik S, Bailly Y, Peyrin JM, Launay JM, Kellermann O, Schneider B. PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer's diseases. Nat Med. 2013 Sep;19(9):1124-31. doi: 10.1038/nm.3302.